A 52-year-old man from Delhi presents with a 3-month history of epigastric pain relieved by food and antacids. He reports black tarry stools for the past 2 weeks and denies NSAID use. On examination, he appears pale with a blood pressure of 100/65 mmHg and heart rate of 102/min. Hemoglobin is 8.2 g/dL. Upper GI endoscopy reveals a 1.5 cm ulcer on the anterior wall of the duodenum with a visible vessel at the base. H. pylori serology is positive. What is the most likely pathophysiologic mechanism underlying ulcer formation in this patient?
A. Increased gastric acid secretion due to H. pylori-induced loss of somatostatin-producing D cells
B. Direct mucosal invasion by H. pylori with subsequent inflammatory cascade and epithelial damage
C. Increased gastrin levels from antral G cells due to hypochlorhydria
D. Decreased mucus production and bicarbonate secretion leading to impaired mucosal defense
Explanation
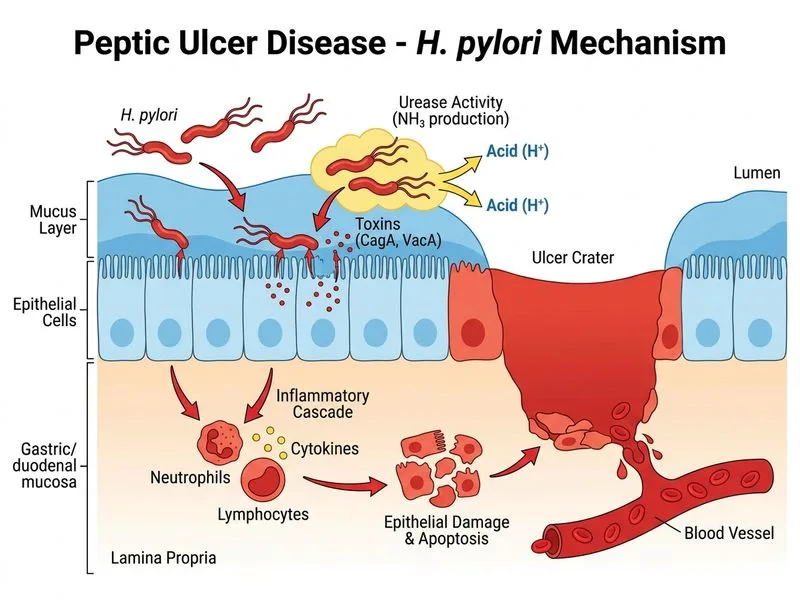
Pathophysiology of H. pylori-Associated Duodenal Ulcer Disease
Primary Mechanism: Loss of D-Cell Somatostatin → Increased Acid Secretion
Key Point
In H. pylori-associated duodenal ulcer disease, the dominant pathophysiologic mechanism is increased gastric acid secretion driven by H. pylori-induced destruction of somatostatin-producing D cells in the antrum.
H. pylori colonizes the gastric antrum and causes:
1.
D-cell loss/dysfunction — Chronic antral inflammation selectively damages somatostatin-producing D cells, removing the principal inhibitory brake on gastrin secretion
2.
Unopposed gastrin release — Antral G cells secrete excess gastrin (hypergastrinemia) in the absence of somatostatin inhibition
Duodenal acid overload — The duodenal mucosa is overwhelmed by excess acid, leading to gastric metaplasia and subsequent H. pylori colonization of the duodenum, culminating in ulceration
Why Duodenal Ulcers Specifically?
Clinical Pearl
Duodenal ulcers are characteristically associated with hyperacidity, in contrast to gastric ulcers which are more often associated with mucosal defense failure. Studies consistently show that patients with H. pylori-positive duodenal ulcers have significantly elevated basal acid output (BAO) and maximal acid output (MAO) compared to controls. Eradication of H. pylori normalizes acid secretion by restoring somatostatin-mediated inhibition of gastrin — confirming that the acid-secretory axis is the primary driver (Harrison's Principles of Internal Medicine, 21e, Ch. 322).
Why the Other Options Are Less Correct
Table
Option
Mechanism
Assessment
A (Correct)
↓ D-cell somatostatin → ↑ gastrin → ↑ acid
Primary mechanism in duodenal H. pylori ulcer
B
↓ Mucus/HCO₃⁻
More relevant to NSAID ulcers and gastric H. pylori ulcers
C
Direct mucosal invasion
H. pylori is non-invasive; it does not penetrate epithelium
D
Hypochlorhydria → ↑ gastrin
Opposite of what occurs in duodenal ulcer (hyperchlorhydria is the rule)
High-YieldNEET PG
Option C is a common distractor — H. pylori is not an invasive organism. It resides in the mucus layer overlying the epithelium and causes damage through urease-generated ammonia, cytotoxins (CagA, VacA), and the resulting inflammatory cascade — but does not directly invade epithelial cells. Option A correctly identifies the net result: loss of D-cell somatostatin → hypergastrinemia → hyperacidity → duodenal ulceration.
Key Point
The classic teaching (Robbins 10e, Ch. 17; KD Tripathi Essentials of Medical Pharmacology) is that H. pylori-associated duodenal ulcers are an acid-hypersecretory state mediated by impaired somatostatin inhibition, distinguishing them mechanistically from gastric ulcers where mucosal defense failure predominates.
Harrison's 21e Ch. 322; Robbins 10e Ch. 17
Loading illustration…
Practice similar questions
Sign up free to access AI-powered MCQ practice with detailed explanations and adaptive learning.