## Clinical Diagnosis: MEN 2A Syndrome ### Defining Triad of MEN 2A **Key Point:** MEN 2A is defined by the triad of **medullary thyroid carcinoma (MTC)**, **pheochromocytoma**, and **primary hyperparathyroidism**. This patient has the first two components with a family history strongly suggestive of the third. ### Clinical Features Matching MEN 2A | Feature | Finding | Significance | |---------|---------|---------------| | **Medullary thyroid carcinoma** | Elevated serum calcitonin (285 pg/mL); brother with MTC; mother died of "thyroid cancer" at 51 | MTC is the most common manifestation (95% of MEN 2A patients); often familial | | **Pheochromocytoma** | Elevated plasma metanephrine (4.2× ULN); hypertension; headaches; 2.8 cm right adrenal mass | Pheochromocytoma occurs in ~50% of MEN 2A patients; causes catecholamine excess | | **Parathyroid involvement** | Normal serum calcium and PTH (currently) | Parathyroid adenoma/hyperplasia occurs in ~20–30% of MEN 2A; may develop later | | **Family history** | Brother with MTC; mother with thyroid cancer (likely MTC); early-onset disease | Autosomal dominant inheritance; RET proto-oncogene mutation | ### Molecular Pathophysiology **High-Yield:** MEN 2A results from **gain-of-function mutations in the RET proto-oncogene** (chromosome 10q11.2). Unlike MEN 1 (loss-of-function), MEN 2 mutations cause constitutive activation of the RET receptor tyrosine kinase, leading to: 1. **C-cell hyperplasia → Medullary thyroid carcinoma** (earliest and most penetrant manifestation) 2. **Chromaffin cell hyperplasia → Pheochromocytoma** (often bilateral; 10% malignant) 3. **Parathyroid hyperplasia → Primary hyperparathyroidism** (least common; may be subclinical) ### Diagnostic Approach **Mnemonic: MEN 2 = **C**alcitonin** — elevated serum calcitonin is the biochemical hallmark. **Clinical Pearl:** In any patient with pheochromocytoma, always screen for MTC (calcitonin) and family history of thyroid cancer. Conversely, in any patient with MTC, screen for pheochromocytoma (plasma metanephrines, imaging) before any intervention to prevent catecholamine crisis. ### Comparison: MEN 2A vs. MEN 2B | Feature | MEN 2A | MEN 2B | |---------|--------|--------| | **RET mutation** | Extracellular/juxtamembrane domain | Intracellular tyrosine kinase domain | | **MTC** | Yes (95%) | Yes (100%, earlier onset) | | **Pheochromocytoma** | Yes (~50%) | Yes (~50%) | | **Parathyroid adenoma** | Yes (~20–30%) | No | | **Mucosal neuromas** | No | Yes (pathognomonic) | | **Marfanoid habitus** | No | Yes | | **Intestinal ganglioneuromatosis** | No | Yes | ### Why Not MEN 1? **Warning:** Do NOT confuse MEN 1 and MEN 2 — they are genetically and clinically distinct. - **MEN 1:** Parathyroid adenoma (95%), gastroenteropancreatic neuroendocrine tumors (60–70%), pituitary adenoma (30–40%). **NO medullary thyroid carcinoma; NO pheochromocytoma.** - **MEN 2A:** Medullary thyroid carcinoma (95%), pheochromocytoma (50%), parathyroid adenoma (20–30%). **NO gastrinomas; NO pituitary adenomas.** This patient has **elevated calcitonin and MTC**, which are **never seen in MEN 1**. ### Management Implications **Key Point:** Patients with MEN 2A require: 1. **RET genetic testing** — confirms diagnosis; identifies at-risk relatives 2. **Prophylactic thyroidectomy** — indicated in childhood (age 5–10) in RET mutation carriers; prevents MTC progression 3. **Adrenal imaging and metanephrine screening** — pheochromocytoma must be excluded before thyroidectomy (risk of hypertensive crisis) 4. **Parathyroid surveillance** — biochemical screening (serum calcium, PTH) every 1–2 years 5. **Family screening** — first-degree relatives should undergo RET testing [cite:Robbins 10e Ch 24; Harrison 21e Ch 439] 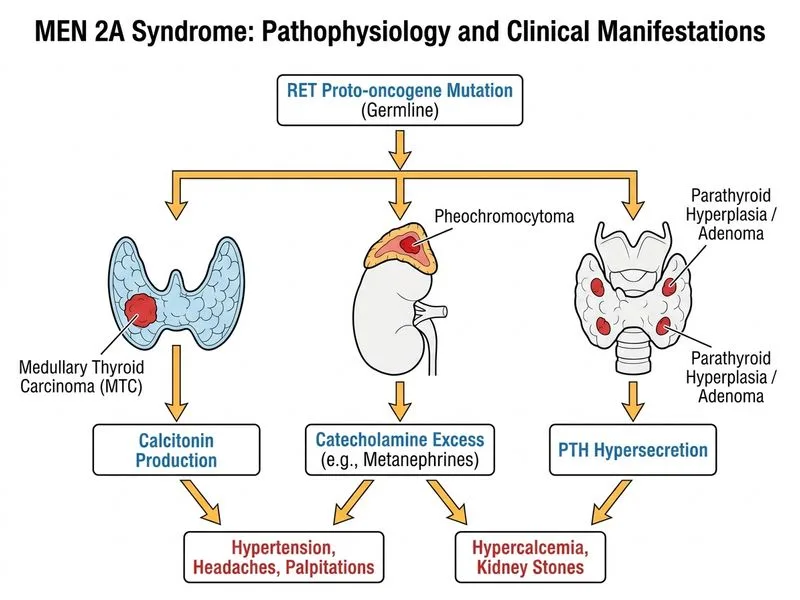
Sign up free to access AI-powered MCQ practice with detailed explanations and adaptive learning.