## RB1 Gene and Retinoblastoma **Key Point:** RB1 (retinoblastoma protein) is the classic tumor suppressor gene associated with hereditary retinoblastoma, inherited in an autosomal dominant pattern with ~90% penetrance. ### Mechanism of Loss 1. RB protein normally acts as a gatekeeper of the G1/S cell cycle checkpoint 2. Hypophosphorylated RB binds E2F transcription factors, preventing S-phase entry 3. Loss of RB function removes this checkpoint control, allowing uncontrolled proliferation ### Clinical Features of RB1 Mutations - **Bilateral retinoblastoma** in ~75% of hereditary cases (vs. ~5% in sporadic) - **Unilateral disease** in ~25% of hereditary cases - **Incomplete penetrance** (~90%) — not all carriers develop tumors - **Increased risk of secondary malignancies** (osteosarcoma, soft tissue sarcoma) especially with radiation therapy ### Two-Hit Hypothesis (Knudson) | Feature | Hereditary | Sporadic | |---------|-----------|----------| | Inheritance | Autosomal dominant | None | | Bilaterality | ~75% | ~5% | | Age of onset | Earlier (mean 18 months) | Later (mean 24 months) | | Number of hits required | 1 (germline) + 1 (somatic) | 2 (both somatic) | **High-Yield:** RB1 mutations predispose to retinoblastoma in childhood; TP53 mutations (Li-Fraumeni) predispose to multiple cancers across all ages. **Clinical Pearl:** Survivors of hereditary retinoblastoma require lifelong surveillance for secondary malignancies, particularly if treated with radiation. 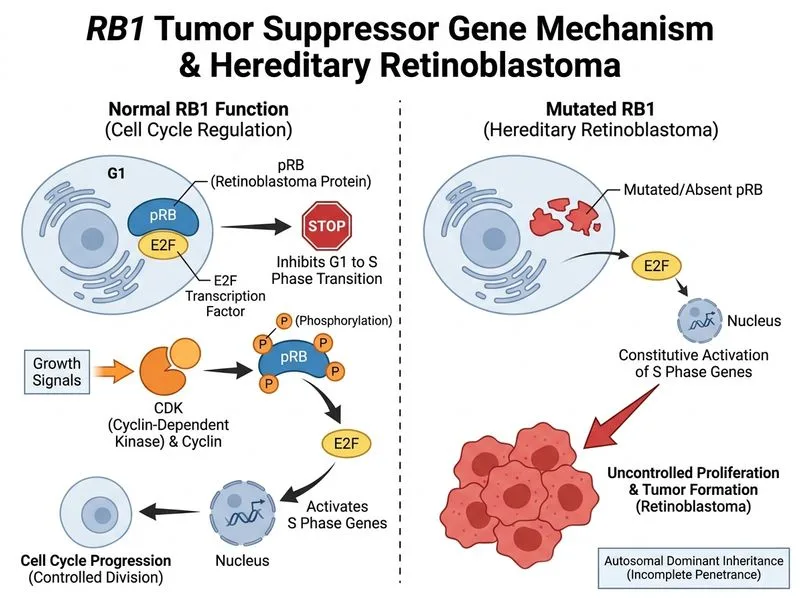
Sign up free to access AI-powered MCQ practice with detailed explanations and adaptive learning.