image mcq
pathology
peripheral blood smear
leukemia
AML M3
CML
platelets
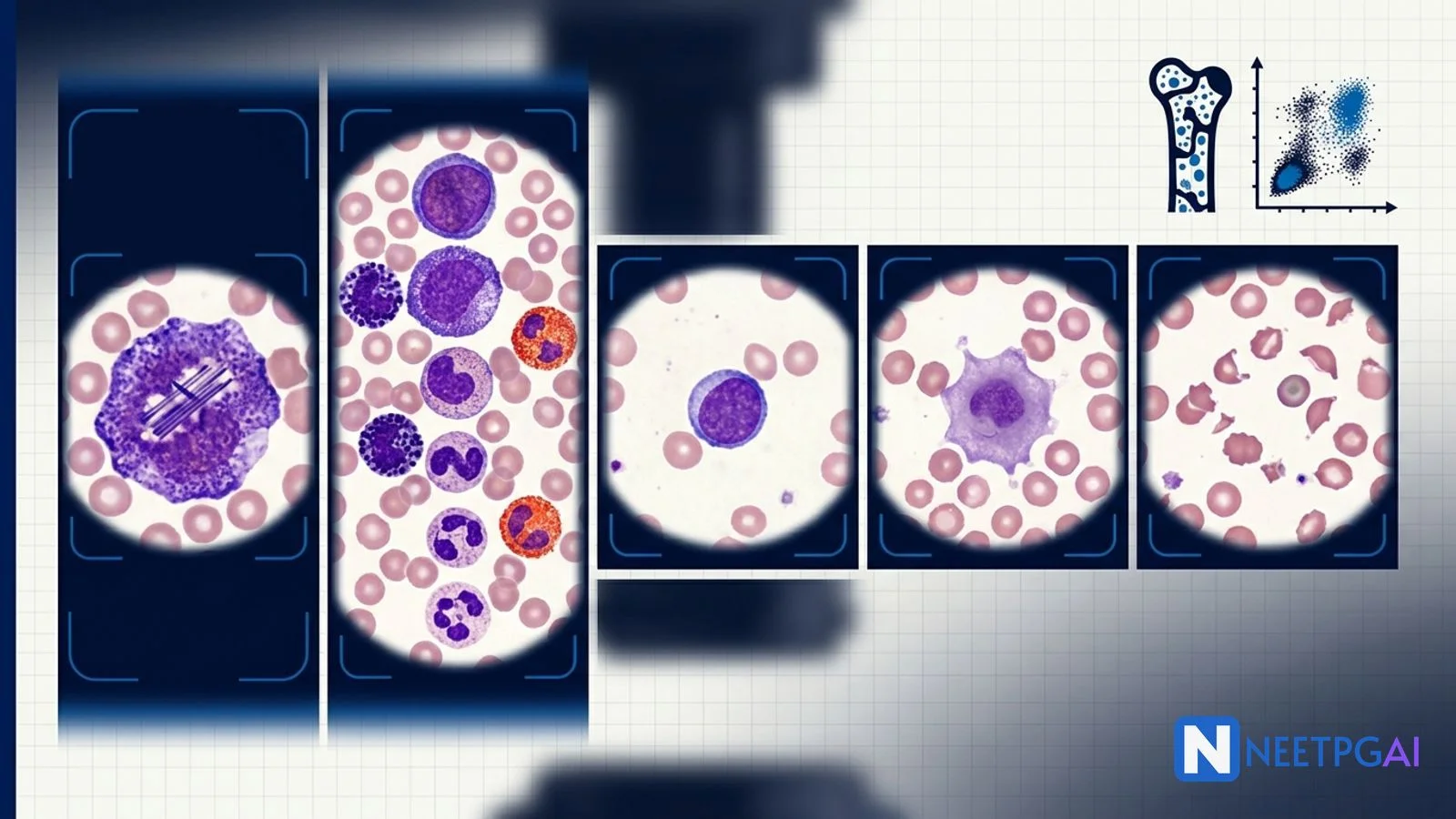
5 high-yield peripheral blood smear image MCQs for NEET PG: AML M3 (APL) Auer rods + faggot cells, CML granulocyte spectrum, ALL lymphoblasts, ITP megathrombocytes, HUS/TTP schistocytes.
Version 1.0 — Published May 2026
Peripheral blood smear interpretation contributes 4-6 questions per NEET PG paper across pathology, haematology, paediatrics, and internal medicine. Five patterns recur reliably:
Combine WBC count + dominant cell + blasts present + platelet adequacy + RBC morphology and almost every NEET PG peripheral blood smear collapses to a 30-second answer.
Peripheral blood smear is the cheapest, fastest, and most diagnostic test in haematology — accessible in every primary health centre across India, often the first investigation that changes the diagnosis from "anaemia" to "leukaemia" or from "fever" to "malaria". NEET PG, INI-CET, and FMGE feature peripheral smear images in 4-6 questions per paper, often coupled with a clinical vignette to test the whole diagnostic chain — pattern recognition, additional confirmatory tests, and management.
The patterns are highly stereotyped. Drilling 5 patterns plus 5-10 PYQ smear images daily for 2 weeks moves accuracy from 40 to 80 percent on this high-yield domain.
| Line | What to look at | Key abnormalities |
|---|---|---|
| Red cells | Size, shape, inclusions, arrangement | Microcytic/macrocytic, schistocyte, target cell, sickle, teardrop, Howell-Jolly, basophilic stippling, parasites |
| White cells | Count estimate, predominant cell, blasts, maturation, dysplasia | Blasts (AML vs ALL), Auer rods, faggot cells, granulocyte maturation spectrum, Pelger-Huet, hypersegmented neutrophils |
| Platelets | Adequate, decreased, increased, morphology | Megathrombocyte, hypogranular, platelet clumping artefact |
This content is for educational purposes for NEET PG exam preparation. It is not a substitute for professional medical advice, diagnosis, or treatment. Clinical information has been reviewed by qualified medical professionals.
Start practicing NEET PG MCQs with AI-powered explanations.
Start Free PracticeMaster GI secretions, digestion, absorption transporters, motility patterns, and gut hormones with high-yield NEET PG 2026 traps and India-context examples.
Master labor stages, Friedman vs Zhang curves, WHO partograph, AMTSL, episiotomy and India JSY/LaQshya policies for NEET PG 2026 OBG MCQs.
5 anterior segment ophthalmology image MCQs for NEET PG: hypopyon and Behcet, Kayser-Fleischer ring in Wilson, Brushfield spots in Down, corneal arcus, and pterygium vs pinguecula.
Daily MCQs, study tips, and topper strategies on Telegram.
Join on Telegram →| Special | Leukoerythroblastic picture, parasites, microorganisms | Nucleated RBCs + immature WBCs together (marrow infiltration), malaria, intracellular bacteria |
| Age | Commonest leukaemia | Key cytogenetics |
|---|---|---|
| Under 1 | Congenital AML, infant ALL with MLL/KMT2A rearrangement | t(4;11), MLL rearrangement (poor prognosis) |
| 2-5 (peak) | ALL (B-cell) | Hyperdiploid (good), TEL-AML1 t(12;21) (good), BCR-ABL t(9;22) (poor) |
| 5-15 | ALL, also AML | Variable |
| 15-30 | ALL, AML, CML, Hodgkin lymphoma | t(15;17) APL, t(8;21) AML, BCR-ABL |
| 30-60 | CML, AML, multiple myeloma | BCR-ABL, FLT3-ITD, NPM1 |
| Over 60 | AML, CLL, multiple myeloma | TP53, complex karyotype |
Image description: [High-power (x1000) Wright-Giemsa stained peripheral blood smear of a 28-year-old woman. Multiple promyelocytes are visible — large cells with abundant granular cytoplasm, an indented or bilobed nucleus, and prominent bundles of needle-shaped Auer rods stacked together (faggot cells). Several promyelocytes contain dozens of Auer rods aggregated in the cytoplasm. There is hypergranular cytoplasm with intense azurophilic granulation. Background red cells appear normal. Platelets are markedly decreased on the smear. WBC count on automated counter is 2,400/μL — leukopenia rather than leukocytosis (the classic 'hypergranular hypocellular' variant of APL).]
Clinical vignette: A 28-year-old previously healthy woman presents with 1 week of progressive fatigue, easy bruising over the arms and thighs, bleeding gums on brushing, and a small epistaxis the previous evening. No fever, no weight loss, no infection. On examination she is pale; multiple ecchymoses on the limbs and trunk; no lymphadenopathy; no hepatosplenomegaly. Labs: Hb 8.2 g/dL, WBC 2,400/μL (neutrophils 35 percent, promyelocytes 45 percent on differential), platelets 18,000/μL. Coagulation: PT 22 sec (raised), aPTT 52 sec (raised), INR 2.0, fibrinogen 0.9 g/L (low), D-dimer markedly raised — overt DIC. Bone marrow aspirate confirms over 80 percent promyelocytes with faggot cells.
Options:
Correct answer: (a) Acute promyelocytic leukaemia (AML M3) with DIC
Reasoning: Young adult with leukopenia or modestly raised WBC, dominance of promyelocytes with bundles of Auer rods (faggot cells), and prominent coagulopathy with overt DIC at presentation is textbook acute promyelocytic leukaemia (APL, AML M3). The cytogenetic hallmark is t(15;17)(q24;q21) producing the PML-RARA fusion gene — confirmed by FISH or PCR. The hypergranular cytoplasm and the bundles of Auer rods discriminate from other AML subtypes.
AML M4 (acute myelomonocytic) produces a mix of myeloblasts and monoblasts with non-specific esterase positivity and gingival hyperplasia — no faggot cells. CML in blast crisis produces a high WBC with a full granulocyte maturation spectrum plus blasts; M3 is leukopenic in 70 percent of cases. ALL produces lymphoblasts without granules or Auer rods, often TdT positive on immunohistochemistry — never faggot cells.
Teaching pearl: APL key features — promyelocytes with faggot cells (bundles of Auer rods), DIC at presentation in 80-90 percent of cases, t(15;17) PML-RARA fusion, leukopenia rather than leukocytosis in 70 percent (hypergranular variant). The hypogranular variant (M3v) has a bilobed/folded nucleus, scant granules, higher WBC, and similar Auer rods on careful examination.
Critical management point — the ATRA emergency:
Differentiator vs other AML subtypes (FAB classification):
| FAB | Description | Hallmark |
|---|---|---|
| M0 | Minimally differentiated | MPO under 3 percent positive, requires immunophenotyping |
| M1 | Without maturation | Blasts predominate, few granules |
| M2 | With maturation | t(8;21) RUNX1-RUNX1T1, Auer rods, good prognosis |
| M3 | Promyelocytic (APL) | t(15;17) PML-RARA, faggot cells, DIC, ATRA + ATO |
| M4 | Myelomonocytic | inv(16) or t(16;16) (M4eo with eosinophilia), good prognosis |
| M5 | Monocytic | t(9;11) MLL, gum hyperplasia, skin infiltration |
| M6 | Erythroid | Dysplastic erythroid precursors |
| M7 | Megakaryoblastic | Down syndrome association in children |
Image description: [High-power Wright-Giemsa peripheral blood smear of a 45-year-old man. There is marked leukocytosis with a full spectrum of granulocyte maturation — myeloblasts (rare, around 2 percent), promyelocytes, myelocytes, metamyelocytes, band forms, and mature neutrophils — all present simultaneously. Basophilia is prominent (15 percent basophils on differential), with eosinophilia (8 percent) also visible. Platelets are increased. Red cells appear normal in number and morphology. No Auer rods are seen in the rare blasts present. The picture is sometimes described as 'all stages of the marrow are in the blood'.]
Clinical vignette: A 45-year-old businessman from Pune presents with 3 months of progressive abdominal fullness, early satiety, weight loss of 4 kg, night sweats, and fatigue. On examination there is massive splenomegaly extending to the umbilicus and beyond into the left iliac fossa, mild hepatomegaly 3 cm below costal margin, and no lymphadenopathy. Labs: Hb 11.2 g/dL, WBC 156,000/μL with the differential described above, platelets 480,000/μL, LDH 1,840 U/L, uric acid 8.4 mg/dL, leukocyte alkaline phosphatase (LAP) score very low (4 — normal 20-100). Bone marrow aspirate shows hypercellular marrow with myeloid hyperplasia and decreased M:E ratio reversed. Cytogenetics show t(9;22)(q34;q11) — Philadelphia chromosome producing the BCR-ABL fusion gene on FISH and PCR.
Options:
Correct answer: (b) Chronic myeloid leukaemia (CML) in chronic phase
Reasoning: Middle-aged adult with massive splenomegaly, WBC over 100,000 with full granulocyte maturation spectrum and prominent basophilia + eosinophilia, low LAP score, and t(9;22) Philadelphia chromosome on cytogenetics is textbook CML in chronic phase. The basophilia is a particularly useful discriminator because it is rarely present in leukemoid reactions.
Leukemoid reaction to infection produces a left shift but typically has fewer immature forms, no basophilia, high LAP score (over 100 — opposite to CML), and a clear inflammatory context. Polycythemia vera presents with raised red-cell mass and JAK2 V617F mutation; the WBC and platelet rise are usually modest. Primary myelofibrosis produces a leukoerythroblastic picture with teardrop cells and bone marrow fibrosis on biopsy, JAK2 or CALR mutations.
Teaching pearl: CML key features — middle-aged adult, massive splenomegaly, full granulocyte maturation in the blood, basophilia + eosinophilia, low LAP score, t(9;22) BCR-ABL Philadelphia chromosome, three phases (chronic → accelerated → blast crisis). Distinguish from leukemoid reaction with the low LAP score + basophilia + BCR-ABL.
Phases of CML:
| Phase | Blasts in marrow/blood | Additional features |
|---|---|---|
| Chronic | Under 10 percent blasts | Most patients present here; well-controlled with TKIs |
| Accelerated | 10-19 percent blasts | Worsening splenomegaly, thrombocytopenia or thrombocytosis unresponsive to therapy, additional cytogenetic abnormalities |
| Blast crisis | Over 20 percent blasts | Behaves like acute leukaemia (AML in 70 percent, ALL in 30 percent), poor prognosis |
Management:
India-specific note: Generic imatinib (Gleevec) is widely available at affordable cost via state TKI programmes and CHAI/Patient Assistance schemes; access has improved 10-year survival in India to approach Western outcomes. Tata Memorial, AIIMS, and CMC Vellore run high-volume CML clinics.
Image description: [High-power Wright-Giemsa peripheral blood smear of a 4-year-old boy. There are numerous small to medium-sized lymphoblasts with scant cytoplasm, condensed nuclear chromatin, and inconspicuous nucleoli (FAB L1 morphology). No Auer rods, no granules in the cytoplasm. Background red cells show mild normocytic anaemia. Platelets are decreased. On flow cytometry (described in vignette) the blasts are TdT positive, CD10 positive (CALLA), CD19 positive, CD22 positive, CD79a positive, MPO negative, surface immunoglobulin negative — common B-cell ALL.]
Clinical vignette: A 4-year-old boy presents with 2 weeks of progressive fatigue, intermittent fever, and complaints of leg pain (he often asks to be carried instead of walking). His mother noticed bruising over the shins and a few petechiae on the trunk. On examination he is pale, has bilateral cervical lymphadenopathy (1-2 cm, firm, non-tender), hepatosplenomegaly (liver 4 cm, spleen 6 cm below costal margin), and ecchymoses on both shins. Labs: Hb 6.8 g/dL, WBC 38,000/μL with 65 percent lymphoblasts on differential, platelets 28,000/μL, LDH 1,420 U/L, uric acid 9.2 mg/dL. Bone marrow aspirate shows 90 percent lymphoblasts replacing normal marrow. Flow cytometry confirms B-cell ALL. Cytogenetics show hyperdiploidy (over 50 chromosomes) — favourable prognosis. CSF cytology shows no leukaemic involvement.
Options:
Correct answer: (a) Acute lymphoblastic leukaemia (B-cell ALL)
Reasoning: Paediatric peak age 2-5 years, fever + bone pain + lymphadenopathy + hepatosplenomegaly + pancytopenia, circulating small lymphoblasts with scant cytoplasm and no Auer rods, and immunophenotype TdT positive + CD10 + CD19 + CD22 + MPO negative is textbook B-cell ALL. Hyperdiploidy carries a favourable prognosis.
AML M2 produces myeloblasts with Auer rods, MPO positive on cytochemistry — different morphology. Burkitt lymphoma produces medium-sized to large blasts with deep blue cytoplasm and vacuoles (starry sky on biopsy), c-myc translocation t(8;14), and FAB L3 morphology. Infectious mononucleosis produces atypical (Downey) lymphocytes with abundant cytoplasm hugging surrounding red cells, not blast cells, and a positive monospot/EBV serology.
Teaching pearl: B-cell ALL key features — paediatric peak 2-5 years, lymphoblasts with scant cytoplasm and condensed chromatin, TdT positive (the discriminator from AML and lymphoma), CD10 positive (CALLA-positive), PAS positive in block pattern, MPO negative. Hyperdiploidy and ETV6-RUNX1 t(12;21) are favourable; Philadelphia chromosome t(9;22) and MLL/KMT2A rearrangement are unfavourable.
Differentiator vs AML:
| Feature | ALL | AML |
|---|---|---|
| Age peak | 2-5 years (paediatric) | Adult, increases with age |
| Cell size | Small to medium | Medium to large |
| Cytoplasm | Scant | More abundant, may have granules |
| Auer rods | Never | May be present (M1, M2, M3) |
| Nucleoli | Inconspicuous (L1), 1-2 prominent (L2) | Prominent |
| MPO | Negative | Positive |
| Sudan black | Negative | Positive |
| TdT | Positive | Negative |
| Flow markers | CD19, CD22, CD79a (B-ALL); CD2, CD3, CD7 (T-ALL) | CD13, CD33, CD117, MPO |
| CNS involvement | Common — requires CNS prophylaxis | Less common |
Management of paediatric B-cell ALL:
Image description: [High-power Wright-Giemsa peripheral blood smear of a 19-year-old woman. The platelet count is markedly decreased — only 1-2 platelets per oil-immersion field. Several megathrombocytes (giant platelets) larger than the surrounding red cells are visible — easily identified by their size (over 4 micrometres, larger than a typical 7-micrometre RBC). Red cells are normal in size, shape, and number. White cells are normal in count and morphology. No schistocytes, no spherocytes, no blasts, no immature granulocytes. The picture is otherwise unremarkable except for the isolated thrombocytopenia with megathrombocytes.]
Clinical vignette: A 19-year-old college student presents with 1 week of easy bruising on the arms and legs and bleeding gums while brushing her teeth. She had a flu-like illness 2 weeks ago that resolved spontaneously. She is otherwise well, not on any medication, no family history of bleeding disorders. On examination: vital signs normal, multiple petechiae on the legs and trunk, ecchymoses on the arms, no lymphadenopathy, no hepatosplenomegaly, no joint swelling, no rash other than petechiae. Labs: Hb 13.2 g/dL (normal), WBC 6,800/μL (normal differential), platelets 8,000/μL (severe thrombocytopenia). LFT, RFT, coagulation (PT, aPTT, fibrinogen) all normal. ANA negative. HIV, HBV, HCV negative. Bone marrow aspirate (done for severe thrombocytopenia): increased number of megakaryocytes, otherwise normal marrow.
Options:
Correct answer: (a) Immune thrombocytopenia (ITP)
Reasoning: Young adult with isolated severe thrombocytopenia, megathrombocytes on smear, no anaemia or leukopenia, no schistocytes, no blasts, normal coagulation, increased marrow megakaryocytes, recent viral illness, and otherwise normal examination and labs is textbook immune thrombocytopenia (ITP, formerly idiopathic thrombocytopenic purpura). The post-viral history and isolated thrombocytopenia with megathrombocytes are highly characteristic.
TTP shows schistocytes (MAHA), thrombocytopenia, neurological symptoms, renal involvement, and very low ADAMTS13 activity — not isolated thrombocytopenia. Aplastic anaemia produces pancytopenia with hypocellular marrow — not increased megakaryocytes. Acute leukaemia would have blasts on smear or in marrow. DIC has prolonged coagulation with low fibrinogen and raised D-dimer — coagulation is normal in ITP.
Teaching pearl: ITP key features — isolated thrombocytopenia (Hb and WBC normal), megathrombocytes on smear (giant platelets reflecting accelerated turnover), increased megakaryocytes in marrow, normal coagulation, often post-viral (especially in children), antiplatelet antibodies (GP IIb/IIIa, GP Ib/IX), diagnosis of exclusion — rule out drug-induced thrombocytopenia, HIV, HCV, SLE, lymphoproliferative disorders.
Pathophysiology:
Management:
ITP in pregnancy: A NEET PG favourite — distinguish from gestational thrombocytopenia (mild, late, no treatment needed) and HELLP/preeclampsia (MAHA, raised LFTs, hypertension). Treat ITP in pregnancy with prednisolone or IVIG; avoid splenectomy in pregnancy if possible.
Image description: [High-power Wright-Giemsa peripheral blood smear of a 6-year-old girl. The dominant abnormality is numerous schistocytes — fragmented red cells, helmet cells, triangular fragments, and small triangular and crescent-shaped fragments — accounting for over 5 percent of red cells. Background red cells show polychromasia (reticulocytosis from compensatory marrow response). Platelets are markedly decreased. White cells are normal in number and morphology. No spherocytes, no target cells, no parasites. The combined picture is microangiopathic hemolytic anaemia (MAHA) + thrombocytopenia.]
Clinical vignette: A 6-year-old girl from Bengaluru is brought by her parents with 1 week of bloody diarrhoea that started after a family barbecue (undercooked beef burger reported), followed by 3 days of pallor, decreased urine output, dark urine, and lethargy. Today she has had no urine output for 18 hours. On examination she is pale, mildly oedematous, mildly hypertensive (BP 124/82 — over 95th centile for age), abdomen mildly distended, no rashes, no hepatosplenomegaly. Labs: Hb 6.4 g/dL, WBC 14,000/μL (mild leukocytosis with normal differential), platelets 28,000/μL, reticulocyte count 8 percent, LDH 1,800 U/L, indirect bilirubin 3.2 mg/dL, haptoglobin undetectable, direct Coombs test NEGATIVE (this is the key — DAT-negative hemolysis), creatinine 4.8 mg/dL (markedly raised), BUN 92, urinalysis shows haematuria and proteinuria. Stool culture grows E. coli O157:H7 producing Shiga-like toxin (Stx-1 and Stx-2) confirmed by PCR. ADAMTS13 activity is 65 percent (normal).
Options:
Correct answer: (a) Hemolytic uraemic syndrome (HUS) — typical, Shiga-toxin-mediated
Reasoning: Child with prodromal bloody diarrhoea following undercooked-beef exposure, classic triad of MAHA (schistocytes, raised LDH, low haptoglobin, indirect hyperbilirubinaemia, DAT-negative) + thrombocytopenia + acute kidney injury, and stool culture growing E. coli O157:H7 with Shiga toxin is textbook typical (post-diarrhoeal, D+) HUS — also called Shiga-toxin-associated HUS or STEC-HUS. The normal ADAMTS13 activity excludes TTP.
TTP shows MAHA + thrombocytopenia + neurological symptoms + renal involvement + fever (classic pentad — full pentad is rare and the modern dyad of MAHA + thrombocytopenia alone is enough to start plasma exchange), and is caused by very low ADAMTS13 (under 10 percent) from autoantibody (acquired) or genetic deficiency (Upshaw-Schulman syndrome). In children, HUS is far more common than TTP. DIC shows prolonged PT/aPTT, low fibrinogen, raised D-dimer — not present in HUS. AIHA shows positive direct Coombs test — DAT is negative in HUS.
Teaching pearl: Typical HUS (STEC-HUS) key features — children 1-10 years, prodromal bloody diarrhoea (E. coli O157:H7 from undercooked beef, unpasteurised milk, contaminated water; less commonly Shigella dysenteriae type 1 in India), MAHA + thrombocytopenia + AKI, DAT-negative hemolysis, normal coagulation, normal ADAMTS13 (the discriminator from TTP). Treatment is supportive — fluid management, dialysis if needed, transfusion if needed, avoid antibiotics (paradoxically increase toxin release in some studies). Self-limiting in 90 percent over 2-3 weeks.
Atypical HUS (aHUS): Complement dysregulation (factor H, factor I, MCP/CD46, factor B, C3 mutations); no diarrhoeal prodrome; chronic relapsing course; treatment with eculizumab (anti-C5 monoclonal antibody).
TTP: MAHA + thrombocytopenia + neurological symptoms + renal involvement + fever (pentad); ADAMTS13 activity under 10 percent; treatment with urgent plasma exchange (PLEX) + steroids + rituximab + caplacizumab. Adults more often than children.
Differentiator vs other causes of MAHA:
| Feature | TTP | Typical HUS (STEC) | Atypical HUS | DIC | Preeclampsia/HELLP |
|---|---|---|---|---|---|
| Age | Adult | Child | Any | Any | Pregnant |
| Prodrome | Variable | Bloody diarrhoea | None | Sepsis, obstetric | Pregnancy |
| Dominant organ | Brain | Kidney | Kidney | Multi-organ | Liver, kidney |
| ADAMTS13 | Under 10% | Normal | Normal | Normal | Normal |
| Coagulation | Normal | Normal | Normal | Prolonged PT/aPTT, low fibrinogen | Variable |
| Coombs | Negative | Negative | Negative | Negative | Negative |
| Treatment | PLEX + steroids + rituximab + caplacizumab | Supportive, dialysis | Eculizumab | Treat cause | Delivery + supportive |
HUS management:
Pitfall 1: Confusing AML M3 with M2. Both have Auer rods, but M3 has bundles of Auer rods (faggot cells), DIC at presentation, t(15;17) PML-RARA, and treated with ATRA + ATO. M2 has single Auer rods, t(8;21) RUNX1-RUNX1T1, and treated with conventional chemotherapy. Missing M3 is life-threatening because of the DIC.
Pitfall 2: Calling every left-shifted leukocytosis 'leukaemia'. Leukemoid reaction to severe infection produces a left shift but typically has higher LAP score, no basophilia, no BCR-ABL, and a clear inflammatory context. CML has low LAP score, basophilia + eosinophilia, BCR-ABL Philadelphia chromosome, and massive splenomegaly.
Pitfall 3: Confusing ALL with AML. Both have circulating blasts, but ALL is TdT positive, MPO negative, smaller cells with scant cytoplasm, no Auer rods, while AML is TdT negative, MPO positive, larger cells with more cytoplasm, may have Auer rods. ALL peaks in children 2-5; AML increases with age.
Pitfall 4: Calling all thrombocytopenia 'ITP'. ITP is isolated thrombocytopenia (Hb and WBC normal) with megathrombocytes, normal coagulation, increased marrow megakaryocytes. Thrombocytopenia with anaemia and schistocytes is MAHA (TTP, HUS, DIC). Thrombocytopenia with leukopenia is bone marrow problem (aplastic anaemia, leukaemia). Thrombocytopenia with prolonged coagulation is DIC.
Pitfall 5: Confusing TTP and HUS. Both produce MAHA + thrombocytopenia. TTP: adult, dominant neurological symptoms, ADAMTS13 under 10 percent, treat with plasma exchange. HUS: child with prodromal bloody diarrhoea, dominant renal involvement, normal ADAMTS13, E. coli O157:H7 toxin, treat supportively. Atypical HUS: complement dysregulation, treat with eculizumab.
Pitfall 6: Missing inherited macrothrombocytopenias. Bernard-Soulier (absent GP Ib/IX/V, giant platelets, family history), May-Hegglin (giant platelets + Dohle-like neutrophil inclusions, autosomal dominant), Wiskott-Aldrich (small platelets, eczema, immunodeficiency, X-linked). Family history and red-cell normality with chronic thrombocytopenia should prompt consideration.
Pitfall 7: Calling a peripheral atypical lymphocyte a 'blast'. Atypical (Downey) lymphocytes in infectious mononucleosis have abundant cytoplasm hugging surrounding red cells, mature chromatin, no Auer rods, no granules — they are reactive T-cells responding to EBV. They are NOT blasts.
Seven recurring patterns. Recognise the pattern and the question collapses to a 30-second answer.
Pattern 1 — The faggot cell question: Smear shows promyelocyte with bundles of Auer rods + young adult with bleeding and DIC. Diagnosis? APL (AML M3). Cytogenetics? t(15;17) PML-RARA. First-line treatment? ATRA + arsenic trioxide; start ATRA empirically before cytogenetic confirmation because DIC is life-threatening.
Pattern 2 — The CML question: Smear shows full granulocyte maturation spectrum + basophilia + middle-aged adult with massive splenomegaly + WBC over 100,000 + low LAP score. Diagnosis? CML chronic phase. Cytogenetics? t(9;22) BCR-ABL Philadelphia chromosome. Treatment? Imatinib (or dasatinib/nilotinib).
Pattern 3 — The ALL question: Smear shows small lymphoblasts + child 2-5 years + fever + bone pain + pancytopenia. Diagnosis? B-cell ALL. Immunophenotype? TdT+, CD10+, CD19+, CD22+, MPO−. Best-prognosis cytogenetics? Hyperdiploidy or ETV6-RUNX1 t(12;21). Worst? BCR-ABL t(9;22) or MLL/KMT2A rearrangement.
Pattern 4 — The ITP question: Smear shows isolated thrombocytopenia with megathrombocytes + normal Hb and WBC + post-viral young patient + no hepatosplenomegaly. Diagnosis? Immune thrombocytopenia (ITP). Bone marrow shows? Increased megakaryocytes. First-line treatment if platelets under 30,000 or bleeding? Prednisolone 1 mg/kg/day.
Pattern 5 — The HUS question: Smear shows schistocytes + thrombocytopenia + child with bloody diarrhoea + AKI + E. coli O157:H7. Diagnosis? Typical HUS (STEC-HUS). ADAMTS13? Normal. Treatment? Supportive, dialysis if needed, AVOID antibiotics and platelet transfusion routinely.
Pattern 6 — The TTP question: Smear shows schistocytes + thrombocytopenia + adult with neurological symptoms + renal involvement + fever. Diagnosis? Thrombotic thrombocytopenic purpura (TTP). ADAMTS13? Under 10 percent. Treatment? Urgent plasma exchange + steroids + rituximab + caplacizumab.
Pattern 7 — The MAHA differential question: Schistocytes on smear. Differential? TTP, HUS (typical and atypical), DIC, preeclampsia/HELLP, malignant hypertension, mechanical heart valve hemolysis, drug-induced MAHA, AIHA-on-MAHA overlap. Coagulation panel and ADAMTS13 are the discriminators.
High-yield one-liners:
Follow a four-line systematic approach for every smear. (1) Red cells — size (normocytic, microcytic, macrocytic), shape (spherocyte, schistocyte, target cell, sickle cell, teardrop, elliptocyte), inclusions (Howell-Jolly, basophilic stippling, Pappenheimer, Heinz, parasites), and arrangement (rouleaux, agglutination). (2) White cells — total count estimate, predominant lineage, blasts present or not (and which type — myeloid vs lymphoid), maturation pattern, dysplasia, granulocyte morphology (Pelger-Huet, hypersegmentation, toxic granules, Dohle bodies, Auer rods). (3) Platelets — adequate, decreased, increased; morphology (megathrombocytes, hypogranular). (4) Special findings — leukoerythroblastic picture (nucleated RBCs + immature WBCs together — indicates marrow infiltration or severe stress), parasites, microorganisms. Then integrate with clinical history and labs.
Auer rods are needle-shaped pink-purple cytoplasmic inclusions composed of fused azurophilic granules, seen in the cytoplasm of myeloblasts in acute myeloid leukaemia. They are pathognomonic for AML (helping distinguish from ALL, where they are never seen). Faggot cells are myeloblasts or promyelocytes containing bundles of multiple Auer rods stacked together resembling a bundle of sticks (faggot). Faggot cells are particularly characteristic of acute promyelocytic leukaemia (AML M3), which has the t(15;17) PML-RARA translocation. M3 is the FAB subtype that responds to all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) and is the only AML subtype where DIC at diagnosis is so common that ATRA is started empirically before cytogenetic confirmation.
Both produce circulating blasts but with distinct morphology and cytochemistry. ALL lymphoblasts are typically smaller, with scant cytoplasm, condensed nuclear chromatin, indistinct nucleoli, NO Auer rods, NO granules. They are TdT-positive on immunohistochemistry, PAS-positive in block pattern (for B-ALL), and myeloperoxidase (MPO) negative. AML myeloblasts are typically larger, with more abundant cytoplasm, fine chromatin, prominent nucleoli, may have Auer rods (M1, M2, M3), may have granules. They are MPO-positive, Sudan black positive, non-specific esterase positive in monocytic subtypes (M4, M5), and TdT negative. Flow cytometry definitively distinguishes — ALL is CD19, CD22, CD79a, TdT positive for B-ALL; CD2, CD3, CD7 for T-ALL. AML is CD13, CD33, CD117, MPO positive. ALL is the commonest paediatric leukaemia (peak age 2-5); AML is commoner in older adults.
Megathrombocytes (also called giant platelets) are platelets larger than a red blood cell — normal platelets are 2-3 micrometres in diameter while megathrombocytes are over 4 micrometres. They reflect increased platelet turnover with release of immature, large platelets from the bone marrow. They appear in (1) immune thrombocytopenia (ITP) — peripheral destruction with compensatory megakaryocyte hyperplasia and release of large young platelets; (2) myeloproliferative disorders (essential thrombocythemia, polycythemia vera, primary myelofibrosis); (3) inherited macrothrombocytopenias (Bernard-Soulier syndrome — giant platelets with absent GP Ib/IX/V, May-Hegglin anomaly — giant platelets with Dohle-like inclusions in neutrophils, MYH9-related disorders, Wiskott-Aldrich syndrome which paradoxically has SMALL platelets); (4) recovery from chemotherapy or thrombocytopenia. Megathrombocytes with thrombocytopenia and otherwise normal smear in an otherwise well child or young adult suggest ITP.
Schistocytes (also called helmet cells, fragmented red cells) are red-cell fragments produced by mechanical shearing through fibrin strands in small vessels. They are the morphological hallmark of microangiopathic hemolytic anaemia (MAHA) and require over 1 percent of red cells fragmented on smear to be considered diagnostic. The differential of MAHA on smear is: (1) thrombotic thrombocytopenic purpura (TTP) — ADAMTS13 deficiency (under 10 percent activity), classic pentad of MAHA + thrombocytopenia + neurological symptoms + renal involvement + fever (full pentad is rare; MAHA + thrombocytopenia alone should trigger urgent plasma exchange); (2) hemolytic uraemic syndrome (HUS) — typically post-diarrhoeal in children with Shiga-toxin E. coli O157:H7 (typical HUS) or atypical HUS from complement dysregulation, dominant renal failure; (3) DIC — sepsis or obstetric causes, prolonged PT/aPTT, low fibrinogen, raised D-dimer (TTP/HUS have normal coagulation); (4) preeclampsia/HELLP; (5) malignant hypertension; (6) mechanical heart valve hemolysis; (7) drug-induced MAHA. Differentiating TTP from HUS and DIC on the smear is impossible — you need ADAMTS13 levels, stool culture, and coagulation panel.
This content is for educational purposes for NEET PG exam preparation. It is not a substitute for professional medical advice, diagnosis, or treatment. Clinical information has been reviewed by qualified medical professionals.
Written by: NEETPGAI Editorial Team Reviewed by: Pending SME Review Last reviewed: May 2026