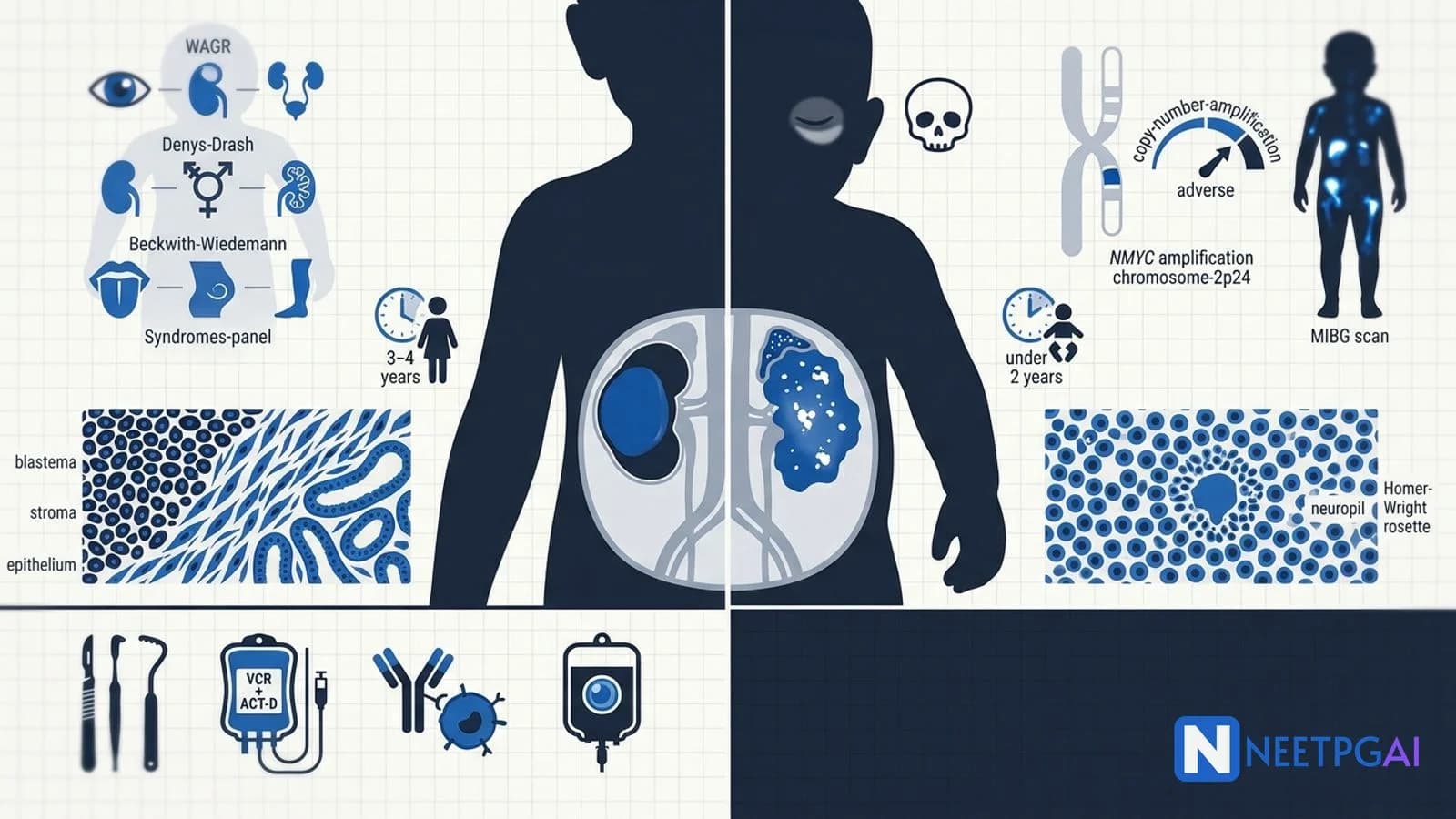
WAGR, Denys-Drash, Beckwith-Wiedemann — three Wilms-predisposing syndromes.
NMYC amplification — strongest adverse prognostic factor in neuroblastoma.
Wilms staging — COG/NWTS I-V; favourable vs unfavourable (anaplastic) histology.
Neuroblastoma staging — INRG L1/L2/M/MS based on image-defined risk factors.
Survival — Wilms greater than 90 percent (FH); neuroblastoma stratified by risk (5-year survival under 50 percent in high-risk).
Wilms tumour and neuroblastoma are the two most common extracranial solid tumours in children, and they together dominate the pediatric oncology section of NEET PG, NEXT and FMGE. The classic exam trap is the "abdominal mass in a toddler" vignette — the examiner expects you to read the precise age, the midline relation, the catecholamine markers and the imaging features and to land on the right diagnosis in under 30 seconds. The 2009 INRG and 2015 ITCC consensuses, plus the updated COG protocols, have shifted some traditional answers, and India's pediatric oncology landscape (AIIMS, Tata Memorial, CMC Vellore, Christian Medical College) has moved toward upfront surgery for resectable Wilms tumours.
This NEETPGAI deep dive covers both tumours head-to-head — pathology, genetics, syndrome associations, staging, treatment, and prognosis — plus a brief comparison to retinoblastoma. Pair this with the colorectal cancer management deep dive and the pediatric imaging image MCQ guide for surgical and radiological coverage.
Wilms tumour (nephroblastoma)
Epidemiology and pathology
The most common renal malignancy in children — about 6 percent of all pediatric cancers. Peak age 3 to 4 years, slightly higher in girls. Arises from persistent nephrogenic rests — small foci of embryonal kidney tissue that should have differentiated but did not. About 10 percent are bilateral (stage V).
Histologically, classic Wilms shows a triphasic pattern — blastema (small blue cells), stroma (mesenchymal), and epithelium (tubular and glomeruloid). Histology is classified as:
Favourable histology (FH) — 90 percent of cases.
Unfavourable histology (UH) — 10 percent; anaplasia (diffuse or focal) — defined by enlarged hyperchromatic nuclei with atypical mitoses. Anaplasia is the single biggest adverse prognostic factor.
Genetics
WT1 gene (11p13) — tumour suppressor required for kidney and gonadal development; mutated in 10 to 20 percent of sporadic Wilms.
WT2 region (11p15) — contains imprinted genes IGF2 and H19; loss of imprinting causes IGF2 overexpression in Beckwith-Wiedemann syndrome and sporadic Wilms.
Predisposing syndromes (high yield)
Syndrome
Genetic basis
Clinical features
WAGR
11p13 deletion (WT1 + PAX6)
Wilms, Aniridia, Genitourinary anomalies, Range of developmental delay
Stage III — Residual tumour confined to abdomen (positive margins, regional lymph nodes, peritoneal spillage during surgery, biopsy before resection).
Stage IV — Haematogenous metastases (lungs commonest, then liver, brain, bone).
Stage V — Bilateral renal involvement at diagnosis.
Treatment
Stage I FH — Nephrectomy plus EE-4A regimen (vincristine + actinomycin D × 18 weeks).
Stage II FH — EE-4A; no radiotherapy.
Stage III FH — Nephrectomy plus DD-4A regimen (vincristine + actinomycin D + doxorubicin × 24 weeks) plus flank radiotherapy.
Stage IV FH — DD-4A plus whole-lung radiotherapy (if pulmonary metastases).
Anaplastic histology — intensified chemotherapy plus radiotherapy regardless of stage.
Stage V (bilateral) — preoperative chemotherapy then nephron-sparing surgery on both sides.
SIOP (European) protocol — preoperative chemotherapy in all cases; used selectively in India for very large unresectable tumours, bilateral disease, or IVC thrombus.
Prognosis — overall 5-year survival greater than 90 percent for FH stage I-III; 80 percent for stage IV FH; 70 percent for anaplastic; 50 percent for stage V.
Neuroblastoma
Epidemiology and pathology
The most common extracranial solid tumour of infancy (under 1 year) and the most common cancer in infants. Median age at diagnosis 17 months; 90 percent diagnosed by age 5.
Histology — sheets of small round blue cells with neuropil and Homer-Wright rosettes (cells around eosinophilic neuropil). Differential diagnosis of "small round blue cell tumours" — neuroblastoma, Ewing sarcoma, rhabdomyosarcoma, lymphoma, retinoblastoma, medulloblastoma.
Genetics and biology
NMYC (MYCN) amplification (2p24) — over 10 copies in 20 percent of cases; strongest single adverse prognostic factor; automatically places tumour in high-risk INRG group.
Chromosome 1p deletion and 11q deletion — adverse.
DNA ploidy — hyperdiploid (good prognosis in infants); diploid/near-diploid (poor).
Dancing eyes, dancing feet (opsoclonus-myoclonus syndrome) — paraneoplastic, occurs in 2 to 3 percent; often the presenting feature; persists after tumour removal.
Blueberry-muffin baby — multiple subcutaneous metastases in infants (stage MS).
Hutchinson syndrome — bone pain, raccoon eyes (skeletal metastases).
Pepper syndrome — massive hepatomegaly from liver metastases in infants (stage MS).
Neuroblastoma origin — neural crest cells; commonest site is adrenal medulla.
NMYC amplification (more than 10 copies, 2p24) — strongest adverse prognostic marker in neuroblastoma.
Urinary VMA and HVA elevated in 90 percent of neuroblastomas — screening test.
Periorbital ecchymoses (raccoon eyes) — orbital bone metastases in neuroblastoma.
Opsoclonus-myoclonus (dancing eyes-dancing feet) — paraneoplastic; can persist after tumour removal.
Homer-Wright rosettes — neuroblastoma; cells around eosinophilic neuropil.
Flexner-Wintersteiner rosettes — retinoblastoma (different — central lumen lined by tumour cells).
MIBG scan — gold standard for neuroblastoma staging and metastasis search.
Stage MS neuroblastoma — infants under 18 months with skin, liver, marrow (under 10 percent) metastases; paradoxically excellent prognosis with frequent spontaneous regression.
Dinutuximab — anti-GD2 monoclonal antibody for high-risk neuroblastoma maintenance.
Catecholamines but NOT hypertensive crisis distinguishes neuroblastoma from phaeochromocytoma (catecholamines are stored chiefly in tumour cells in NB).
Recent updates and Indian context
InPOG-Wilms tumour study — Indian Paediatric Oncology Group multi-centre protocol that documented over 90 percent overall survival in FH Wilms across 8 Indian centres (AIIMS Delhi, Tata Memorial Mumbai, CMC Vellore, RMH Hyderabad, KIMS Bangalore).
Indian neuroblastoma cohort data — over 60 percent present with high-risk metastatic disease (worse than Western cohorts) reflecting delayed presentation; AIIMS protocol focuses on intensified induction and improved supportive care.
National Cancer Grid (NCG) protocols — standardised paediatric oncology pathways across over 250 Indian cancer centres; available free at ncg.org.in.
PMJAY (Ayushman Bharat) — covers paediatric oncology treatment including dinutuximab and autologous stem cell transplant since 2021.
GD2-CAR-T cell therapy — under clinical trial for relapsed neuroblastoma in select Indian centres.
Targeted ALK inhibitors (lorlatinib) — newer high-risk neuroblastoma rescue agent; AIIMS using on compassionate basis.
131I-MIBG therapy — increasingly available at major Indian nuclear-medicine centres for refractory neuroblastoma.
Frequently asked questions
How do you clinically distinguish Wilms tumour from neuroblastoma?
Both present as abdominal mass in a young child but several features separate them. Wilms tumour peaks at 3 to 4 years, usually does NOT cross the midline (renal-bound, smooth and well-encapsulated), causes hypertension and haematuria, and rarely metastasises at presentation. Neuroblastoma peaks under 2 years, typically CROSSES the midline (irregular and calcified on imaging), causes catecholamine excess (sweating, tachycardia), periorbital ecchymoses (raccoon eyes from skull metastases), opsoclonus-myoclonus, and over 60 percent have metastases at diagnosis.
What is the genetic basis of Wilms tumour and which syndromes are associated?
Wilms tumour arises from persistent nephrogenic rests. WT1 gene at 11p13 (a tumour suppressor required for normal kidney and gonadal development) is mutated in about 20 percent of cases. WT2 region at 11p15 (containing imprinted genes IGF2 and H19) is the second locus. Three syndromes: WAGR (Wilms, Aniridia, Genitourinary anomalies, Range of developmental delay — 11p13 deletion); Denys-Drash (Wilms, intersex disorders, progressive nephropathy — WT1 missense); Beckwith-Wiedemann (macroglossia, organomegaly, hemihypertrophy, omphalocele — 11p15 imprinting defect).
Why is NMYC amplification important in neuroblastoma?
NMYC (now MYCN) is a proto-oncogene on chromosome 2p24. Amplification (more than 10 copies) is found in about 20 percent of neuroblastomas and is the strongest single adverse prognostic marker — it automatically places the tumour in the high-risk INRG category regardless of stage or age. NMYC-amplified tumours respond initially to chemotherapy but relapse rapidly. Treatment intensifies to include high-dose chemotherapy with autologous stem cell rescue, surgery, radiation, 13-cis-retinoic acid, and anti-GD2 immunotherapy (dinutuximab).
What is the COG/NWTS staging system for Wilms tumour?
The Children's Oncology Group (COG, formerly NWTS — National Wilms Tumor Study) stages Wilms tumour by anatomical extent. Stage I — tumour limited to kidney, completely resected. Stage II — extends beyond kidney but completely resected (vascular or capsular invasion). Stage III — residual tumour confined to abdomen (positive margins, lymph node involvement, peritoneal spillage). Stage IV — distant metastases (lungs, liver, brain). Stage V — bilateral renal involvement at diagnosis (5 percent). Histology is independently classified as favourable (FH, 90 percent) or unfavourable (UH, 10 percent — anaplastic), which crucially modifies treatment intensity and prognosis.
What is the management approach for Wilms tumour in India?
The Indian standard is upfront radical nephrectomy when feasible (COG approach) — laparotomy through a transabdominal incision with full abdominal exploration, ligation of the renal vessels first, en bloc resection of the kidney and tumour, and sampling of regional lymph nodes. SIOP (European) preoperative chemotherapy is used in bilateral disease (stage V) and very large tumours where nephrectomy is unsafe. Post-operative therapy is staged-and-histology-based — stage I FH gets vincristine plus actinomycin (EE-4A regimen); higher stages add doxorubicin and radiation. Overall survival exceeds 90 percent for favourable histology stage I-III. AIIMS and Tata Memorial Centre lead protocols nationally.
This content is for educational purposes for NEET PG exam preparation. It is not a substitute for professional medical advice, diagnosis, or treatment. Clinical information has been reviewed by qualified medical professionals.
Written by: NEETPGAI Editorial Team
Reviewed by: Pending SME Review
Last reviewed: May 2026