Quick Answer
Biochemistry contributes 4 to 6 NEET PG questions per paper, weighted toward metabolic pathways and inborn errors of metabolism (IEM). Lock these:
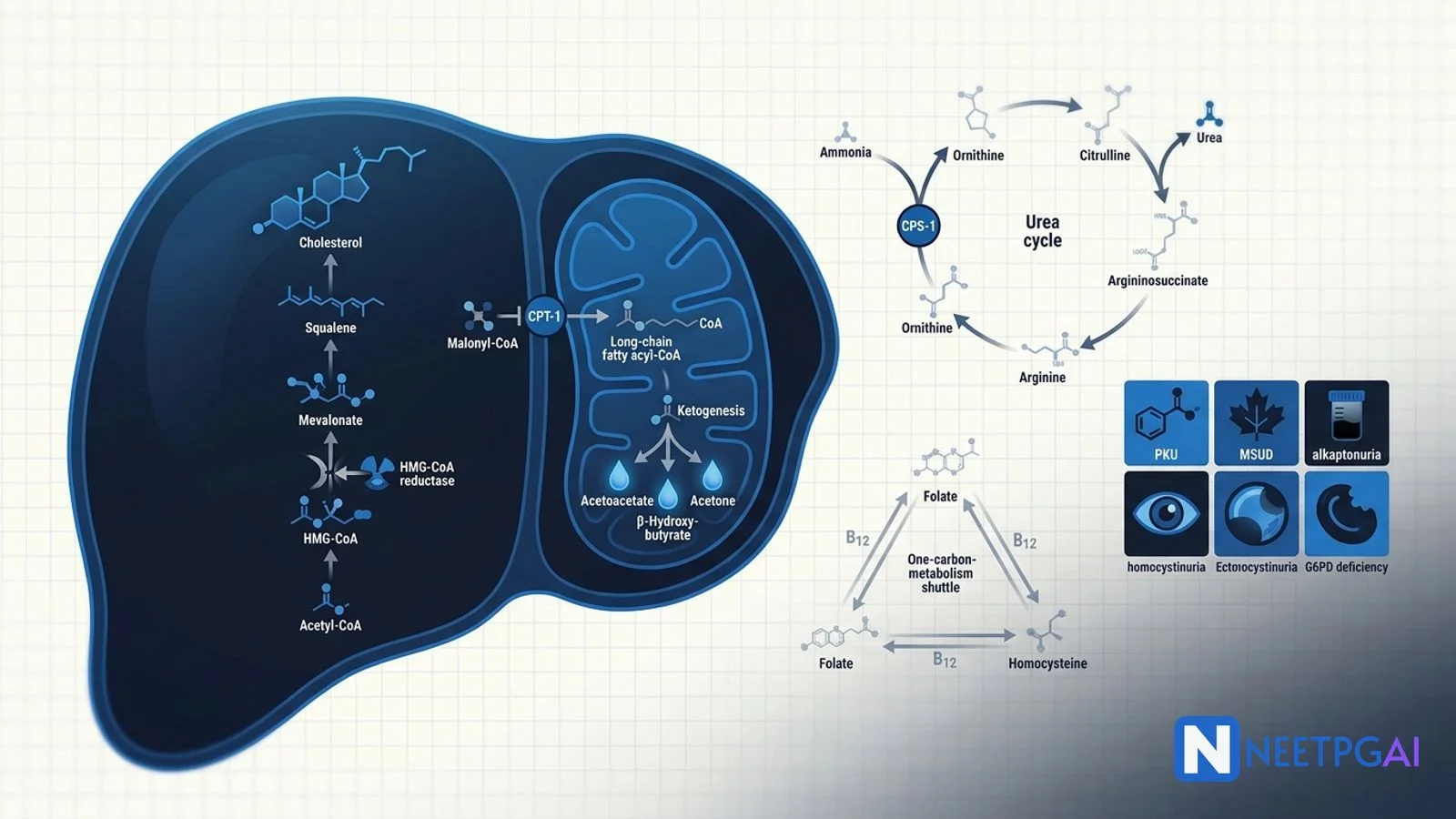
- Cholesterol biosynthesis — HMG-CoA reductase rate-limiting; statins block it.
- Beta-oxidation — carnitine shuttle (CPT-1, CPT-2); produces acetyl-CoA, NADH, FADH2.
- Ketogenesis — hepatic only; acetoacetate, beta-hydroxybutyrate, acetone; DKA mechanism.
- Urea cycle — CPS-1 rate-limiting; OTC X-linked; hyperammonaemia + respiratory alkalosis.
- One-carbon metabolism — folate, B12, methionine, homocysteine.
- IEM core five — PKU, MSUD, alkaptonuria, homocystinuria, galactosemia.
- G6PD deficiency — X-linked; NADPH/glutathione; haemolysis with oxidant stress.
Biochemistry is the most "decodable" NEET PG subject — every pathway has one rate-limiting enzyme, one regulatory pivot, and one clinical correlate that examiners ride. This NEETPGAI guide combines lipid and amino acid metabolism with the inborn errors that dominate paediatric, medicine and pathology vignettes. The reward for systematic study is disproportionate: a single afternoon on rate-limiting enzymes can lock five marks.
Pair this deep dive with the carbohydrate metabolism guide and the enzyme kinetics primer for a complete metabolic map.
Lipid metabolism
Cholesterol biosynthesis
All 27 carbons come from acetyl-CoA. The pathway is cytoplasmic with mitochondrial citrate shuttle providing acetyl-CoA.
- Acetyl-CoA + acetoacetyl-CoA → HMG-CoA (HMG-CoA synthase, cytosolic).
- HMG-CoA → mevalonate — HMG-CoA reductase. Rate-limiting. Blocked by statins. Regulated by SREBP (sterol-responsive element-binding protein) — high cholesterol blocks SREBP cleavage and shuts down the enzyme.
- Mevalonate → isopentenyl-PP → squalene → lanosterol → cholesterol.
Cholesterol is the precursor for bile acids (liver), steroid hormones (adrenal, gonad), vitamin D (skin), and a structural component of membranes.
Lipoproteins
Five classes, decreasing density: chylomicrons (dietary triglycerides from gut), VLDL (hepatic triglycerides), IDL, LDL (cholesterol delivery to peripheral cells), HDL (reverse cholesterol transport from periphery to liver).
Key enzymes — lipoprotein lipase (LPL) on endothelium hydrolyses chylomicron and VLDL triglycerides (deficiency = familial chylomicronaemia, eruptive xanthomas, pancreatitis); lecithin-cholesterol acyltransferase (LCAT) esterifies cholesterol on HDL; CETP exchanges cholesterol ester from HDL for triglyceride from VLDL; PCSK9 degrades hepatic LDL receptors (PCSK9 inhibitors raise LDL clearance).
Fatty acid oxidation
Long-chain fatty acids enter mitochondria via the carnitine shuttle:
- Acyl-CoA synthetase activates fatty acid to fatty acyl-CoA on the outer membrane.
- CPT-1 (outer membrane) swaps CoA for carnitine. Rate-limiting, inhibited by malonyl-CoA (the first committed metabolite of fatty acid synthesis).
- Carnitine acyl-translocase moves acyl-carnitine across the inner membrane.
- CPT-2 (matrix) swaps carnitine back for CoA.
Once in the matrix, beta-oxidation spirals — each turn yields acetyl-CoA, NADH and FADH2. Each acetyl-CoA enters the TCA cycle (12 ATP). A 16-carbon palmitate yields 106 ATP net.
Defects — MCAD deficiency (medium-chain acyl-CoA dehydrogenase) causes hypoketotic hypoglycaemia in infants after fasting, with elevated C8-C10 acylcarnitines on tandem MS newborn screening. Carnitine deficiency mimics MCAD with cardiomyopathy.
Ketogenesis
Hepatic mitochondria only. Acetyl-CoA from beta-oxidation cannot enter TCA when oxaloacetate is diverted to gluconeogenesis (fasting, DKA). Two acetyl-CoA → acetoacetyl-CoA → HMG-CoA → acetoacetate → beta-hydroxybutyrate (predominant in DKA, ratio 3:1 over acetoacetate) and acetone (volatile, fruity breath).
Ketone bodies are used by brain, heart and skeletal muscle. The liver lacks succinyl-CoA-acetoacetate-CoA transferase (thiophorase) and cannot use them.
In DKA, insulin deficiency unbridles hormone-sensitive lipase → free fatty acids flood the liver → beta-oxidation → acetyl-CoA → ketones (high anion gap metabolic acidosis). Treat with insulin, IV fluids, potassium replacement, and identify trigger.
Fatty acid synthesis
Cytoplasmic, opposite of beta-oxidation. Acetyl-CoA leaves mitochondria as citrate via the citrate shuttle. Acetyl-CoA carboxylase (ACC) converts acetyl-CoA to malonyl-CoA — the rate-limiting step, biotin-dependent, activated by citrate and insulin, inhibited by palmitoyl-CoA and glucagon. Fatty acid synthase (a multi-enzyme complex) adds two-carbon units from malonyl-CoA to a growing chain, using NADPH from the PPP. Final product = palmitate (16C).
Amino acid metabolism
Transamination
Amino group transfer from amino acid to alpha-ketoglutarate forming glutamate and a new alpha-keto acid. Catalysed by aminotransferases (ALT and AST), pyridoxal phosphate (vitamin B6) cofactor.
- ALT (alanine aminotransferase) — liver-specific; rises in hepatocellular injury.
- AST (aspartate aminotransferase) — also in heart, muscle, RBC. AST:ALT ratio greater than 2 suggests alcoholic liver disease (B6 deficiency limits ALT more than AST).
Glutamate then surrenders its amino group as ammonia via glutamate dehydrogenase (the only enzyme that uses NAD+ or NADP+).
Urea cycle
Five enzymes; two mitochondrial (CPS-1, OTC) and three cytosolic. The matrix nitrogen comes from ammonia via CPS-1 (carbamoyl phosphate synthetase 1, rate-limiting, activated by N-acetylglutamate which is high when arginine is plentiful — protein meal). The cytosolic nitrogen comes from aspartate.
Steps:
- NH3 + CO2 + 2 ATP → carbamoyl phosphate (CPS-1, mitochondria).
- Carbamoyl phosphate + ornithine → citrulline (OTC, mitochondria).
- Citrulline + aspartate + ATP → argininosuccinate (cytosol).
- Argininosuccinate → arginine + fumarate.
- Arginine + H2O → urea + ornithine.
Defects (all autosomal recessive except OTC, which is X-linked) present with hyperammonaemic encephalopathy, respiratory alkalosis (NH3 stimulates respiratory centre), vomiting, lethargy, coma. Triggered by protein load, infection or catabolic stress. Treatment — protein restriction, sodium benzoate/phenylacetate (alternate nitrogen disposal), arginine supplementation (except in arginase deficiency), dialysis if severe.
One-carbon metabolism
Folate carries one-carbon units (methylene, methyl, formyl) for nucleotide synthesis. Vitamin B12 (methylcobalamin) transfers a methyl group from N5-methyl-THF to homocysteine → methionine (methionine synthase) and regenerates THF — the "folate trap" in B12 deficiency: all folate is stuck as N5-methyl-THF and cannot be used for DNA synthesis, mimicking folate deficiency.
Homocysteine has two fates:
- Remethylation to methionine (methionine synthase, B12).
- Transsulfuration to cysteine (cystathionine beta-synthase, B6).
Deficiencies of B6, B9, B12 cause hyperhomocysteinaemia, a cardiovascular and thrombosis risk factor. MTHFR C677T polymorphism is the commonest cause of mild hyperhomocysteinaemia in the Indian population.
Inborn errors of metabolism — the core five
Phenylketonuria (PKU)
- Enzyme — phenylalanine hydroxylase (BH4 cofactor).
- Inheritance — autosomal recessive.
- Accumulates — phenylalanine, phenylpyruvate, phenylacetate, phenyllactate.
- Deficient — tyrosine (precursor of melanin, dopamine, thyroid hormone).
- Presents — normal at birth; by 6 months intellectual disability, eczema, hypopigmentation, musty mousy odour, seizures.
- Screening — Guthrie card (Bacillus subtilis growth assay) or tandem MS.
- Treatment — lifelong low-phenylalanine diet (avoid aspartame); sapropterin (synthetic BH4) for BH4-responsive variants.
- Maternal PKU — fetal anomalies (microcephaly, congenital heart disease) even in non-PKU fetus if mother has elevated phenylalanine.
Maple syrup urine disease (MSUD)
- Enzyme — branched-chain alpha-ketoacid dehydrogenase (BCKDH).
- Accumulates — branched-chain amino acids (leucine, isoleucine, valine) and their keto acids.
- Presents — neonatal vomiting, hypertonia, seizures, opisthotonos, urine smells like maple syrup or burnt sugar.
- Treatment — dietary restriction of BCAA, thiamine (cofactor of BCKDH), peritoneal dialysis if acute.
Alkaptonuria
- Enzyme — homogentisate oxidase.
- Accumulates — homogentisic acid.
- Presents — urine darkens on standing (alkali-induced oxidation); ochronosis (blue-black pigmentation of cartilage, sclera); arthritis in adulthood.
- Treatment — nitisinone, vitamin C, low-protein diet.
Homocystinuria
- Enzyme — cystathionine beta-synthase (CBS), B6-dependent.
- Accumulates — homocysteine, methionine.
- Presents — Marfanoid habitus, downward lens dislocation (Marfan = upward), intellectual disability, premature atherosclerosis and thrombosis, osteoporosis.
- Treatment — pyridoxine (B6) for responsive variants, low-methionine diet, betaine.
Galactosemia (classic)
- Enzyme — galactose-1-phosphate uridyltransferase (GALT).
- Accumulates — galactose-1-phosphate, galactitol.
- Presents — failure to thrive after milk feeds, vomiting, jaundice, hepatomegaly, E. coli sepsis, cataracts (galactitol in lens), intellectual disability if untreated.
- Treatment — lifelong galactose- and lactose-free diet.
G6PD deficiency (honourable mention)
- Enzyme — glucose-6-phosphate dehydrogenase (PPP, rate-limiting).
- Inheritance — X-linked recessive.
- Mechanism — NADPH deficiency → glutathione cannot be reduced → oxidant stress oxidises haemoglobin (Heinz bodies, bite cells) and ruptures RBCs.
- Triggers — fava beans, primaquine, sulpha drugs, nitrofurantoin, dapsone, infection, naphthalene.
- Presents — episodic intravascular haemolysis (haemoglobinuria, jaundice) after exposure.
- Diagnosis — fluorescent spot test, quantitative G6PD enzyme assay (do NOT test during acute haemolysis — reticulocytes have normal enzyme, false-negative).
NEET PG MCQ traps
- HMG-CoA reductase — cholesterol rate-limiting; statins.
- CPT-1 — beta-oxidation rate-limiting; inhibited by malonyl-CoA.
- Acetyl-CoA carboxylase (ACC) — fatty acid synthesis rate-limiting; biotin-dependent.
- CPS-1 — urea cycle rate-limiting; activated by N-acetylglutamate.
- OTC deficiency — only X-linked urea cycle defect.
- Hyperammonaemia + respiratory alkalosis = urea cycle defect.
- Hyperammonaemia + metabolic acidosis = organic acidurias (propionic, methylmalonic).
- Maternal PKU affects fetus regardless of fetal genotype.
- Downward lens dislocation = homocystinuria (Marfan = upward).
- Methylmalonyl-CoA mutase requires B12; deficiency causes methylmalonic acidaemia.
- DKA dominant ketone = beta-hydroxybutyrate (acetoacetate reduced; ratio 3:1).
- Bite cells and Heinz bodies = G6PD deficiency.
- Folate trap in B12 deficiency — all folate stuck as N5-methyl-THF.
- Galactosemia infant + E. coli sepsis + cataracts — classic vignette.
- MCAD deficiency — hypoketotic hypoglycaemia in infants; C8-C10 acylcarnitines on tandem MS.
Recent updates and Indian context
- National Rural Health Mission (NRHM) and the National Newborn Screening pilot (NEDP) are expanding tandem-MS-based screening for IEM in tertiary centres. Punjab, Kerala, Tamil Nadu, Goa and Chandigarh have universal newborn screening programmes; many other states are partial.
- G6PD deficiency has high prevalence in the Indian tribal belt; primaquine for vivax malaria mandates G6PD screening per NVBDCP 2024 guidelines.
- MTHFR C677T polymorphism — prevalent in north Indian populations; clinical significance for thrombosis is contested but features in NEET PG vignettes.
- PCSK9 inhibitors (alirocumab, evolocumab) and inclisiran (siRNA against PCSK9) are now in Indian guidelines for refractory familial hypercholesterolaemia.
Frequently asked questions
What is the rate-limiting enzyme of cholesterol biosynthesis and how is it regulated?
HMG-CoA reductase is the rate-limiting enzyme. It is regulated by sterol-mediated feedback (cholesterol suppresses SREBP processing), insulin (activates dephosphorylated active form), glucagon and AMPK (phosphorylate the inactive form), and pharmacologically inhibited by statins. Statins competitively block HMG-CoA reductase, deplete intracellular cholesterol, upregulate LDL receptors and lower serum LDL by up to 50 percent.
Why is the carnitine shuttle essential for fatty acid oxidation?
Long-chain fatty acyl-CoA cannot cross the inner mitochondrial membrane. CPT-1 on the outer membrane swaps CoA for carnitine to form acyl-carnitine, which is transported in by carnitine acyl-translocase, and CPT-2 on the matrix side swaps carnitine back for CoA. CPT-1 is inhibited by malonyl-CoA (the first committed intermediate of fatty acid synthesis), preventing simultaneous synthesis and oxidation — a classic reciprocal regulation.
Why is the urea cycle the key disposal route for ammonia?
Free ammonia is neurotoxic. The urea cycle (Krebs-Henseleit cycle) condenses two nitrogens — one from carbamoyl phosphate (CPS-1, the rate-limiting mitochondrial enzyme, allosterically activated by N-acetylglutamate) and one from aspartate — into urea, which is water-soluble and renally excreted. Defects (OTC deficiency is the commonest, X-linked) present with hyperammonaemic encephalopathy and respiratory alkalosis triggered by protein loads or catabolic stress.
What is the biochemical basis of phenylketonuria?
PKU is autosomal recessive deficiency of phenylalanine hydroxylase, which converts phenylalanine to tyrosine using BH4. Phenylalanine accumulates and is shunted to phenylpyruvate, phenylacetate and phenyllactate, causing intellectual disability, eczema, a musty mousy odour and hypopigmentation (tyrosine is the precursor of melanin). Treatment is lifelong low-phenylalanine diet; sapropterin (BH4) helps responsive variants. Newborn screening with the Guthrie card or tandem MS prevents disability.
Why does G6PD deficiency cause haemolysis?
G6PD is the rate-limiting enzyme of the pentose phosphate pathway, generating NADPH. NADPH keeps glutathione reduced — glutathione neutralises hydrogen peroxide and other oxidants. In G6PD deficiency, oxidative stress (fava beans, primaquine, sulpha drugs, infection) overwhelms reduced glutathione, oxidises haemoglobin to methaemoglobin and crosslinks it to membrane (Heinz bodies), causing intravascular and extravascular haemolysis. Bite cells appear on the smear.
This content is for educational purposes for NEET PG exam preparation. It is not a substitute for professional medical advice, diagnosis, or treatment. Clinical information has been reviewed by qualified medical professionals.
Written by: NEETPGAI Editorial Team
Reviewed by: Pending SME Review
Last reviewed: May 2026